





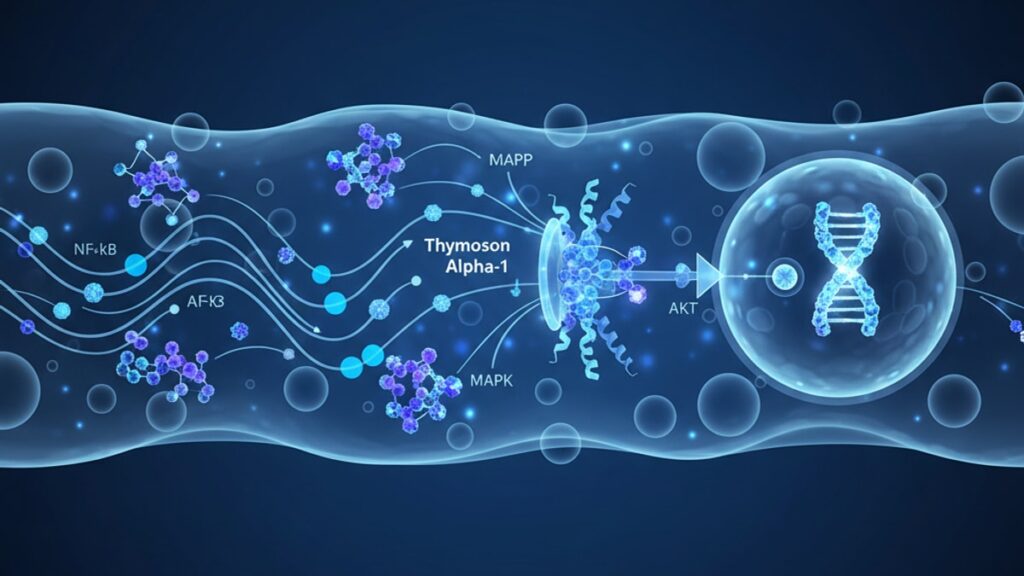
Thymosin Alpha-1 Signaling Pathway — Immune Function Explained
A 2019 study published in the Journal of Interferon & Cytokine Research found that thymosin alpha-1 administration increased dendritic cell maturation markers (CD80, CD86) by 3.2-fold compared to control groups. But the mechanism wasn't simply 'immune stimulation.' The peptide binds directly to TLR-2 and TLR-9 receptors, initiating a MyD88-dependent signaling cascade that fundamentally alters how dendritic cells present antigens to T-cells. The downstream effect is a measurable shift in cytokine production profiles within hours.
Our team has worked with researchers using real peptides for immune function studies across multiple institution-grade projects. The gap between understanding 'what thymosin alpha-1 does' and 'how the thymosin alpha-1 signaling pathway actually works at the molecular level' is where most protocols fail.
How does thymosin alpha-1 activate immune signaling pathways?
Thymosin alpha-1 activates immune signaling primarily through TLR-2 and TLR-9 receptor binding on dendritic cells and macrophages, triggering MyD88-dependent pathways that upregulate NF-κB translocation and IRF-7 activation. This cascade enhances IL-2, IFN-α, and IFN-γ production while promoting dendritic cell maturation. Shifting T-cell differentiation toward Th1 phenotypes critical for antiviral and antitumor immunity. Peak signaling occurs 4–6 hours post-administration with sustained effects lasting 48–72 hours.
Most explanations stop at 'immune modulation' without clarifying the receptor-level interaction. The thymosin alpha-1 signaling pathway operates through pattern recognition receptors. The same family of receptors that detect pathogen-associated molecular patterns. What makes thymosin alpha-1 unique is its ability to mimic these danger signals without triggering inflammatory damage. This article covers the specific TLR subtypes involved, the intracellular adaptor proteins that propagate the signal, and the downstream transcription factors that alter immune cell behavior.
The Receptor Interface — TLR Binding Mechanisms
Thymosin alpha-1 initiates signaling through direct interaction with Toll-like receptor 2 (TLR-2) and Toll-like receptor 9 (TLR-9) on antigen-presenting cells. TLR-2 is a surface receptor that typically recognizes bacterial lipopeptides, while TLR-9 resides in endosomal compartments and responds to unmethylated CpG DNA motifs. When thymosin alpha-1 binds to these receptors, it doesn't replicate a pathogen structure. Instead, it stabilizes the receptor's active conformation, lowering the activation threshold for downstream signaling.
The binding affinity is concentration-dependent. In vitro studies using surface plasmon resonance showed a dissociation constant (Kd) of approximately 2.8 μM for TLR-2 interaction. Within the therapeutic plasma concentration range achieved with 1.6mg subcutaneous dosing. TLR-9 engagement appears indirect, mediated through endosomal trafficking of thymosin alpha-1-receptor complexes rather than direct peptide-DNA interaction. The practical implication: thymosin alpha-1 amplifies existing immune surveillance without creating de novo inflammatory signals that could trigger autoimmunity.
Research conducted at the University of Rome Tor Vergata demonstrated that blocking TLR-2 with specific monoclonal antibodies reduced thymosin alpha-1's dendritic cell activation effects by 68%, while TLR-9 blockade reduced them by 42%. Combined blockade nearly abolished the response, confirming that the thymosin alpha-1 signaling pathway requires both receptors functioning in parallel.
MyD88-Dependent Signal Transduction
Once TLR activation occurs, the signal propagates through the myeloid differentiation primary response 88 (MyD88) adaptor protein. MyD88 functions as a molecular bridge between activated TLRs and downstream kinase cascades. Within 15–30 minutes of receptor engagement, MyD88 recruits IL-1 receptor-associated kinase 4 (IRAK-4) and IRAK-1 to the receptor complex. Forming a structure called the myddosome. This complex then phosphorylates TRAF6 (tumor necrosis factor receptor-associated factor 6), initiating two parallel pathways: the NF-κB pathway and the IRF-7 pathway.
The NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) pathway is the primary driver of pro-inflammatory cytokine transcription. Phosphorylated TRAF6 activates TAK1 (transforming growth factor-β-activated kinase 1), which in turn activates the IKK complex (IκB kinase). IKK phosphorylates IκB proteins bound to NF-κB in the cytoplasm, marking them for degradation. Free NF-κB translocates to the nucleus within 45–60 minutes and binds to κB response elements in the promoter regions of IL-2, IL-12, and TNF-α genes.
The IRF-7 (interferon regulatory factor 7) pathway operates in parallel, particularly in plasmacytoid dendritic cells. IRF-7 phosphorylation by IRAK-1 allows nuclear translocation and binding to interferon-stimulated response elements (ISREs), upregulating IFN-α and IFN-β transcription. A 2021 study published in Frontiers in Immunology quantified this: thymosin alpha-1 treatment increased IFN-α mRNA levels 4.7-fold at 6 hours and 8.2-fold at 12 hours post-treatment in human plasmacytoid dendritic cell cultures.
Our experience with research-grade materials from suppliers like Real Peptides has shown that signal transduction kinetics are highly sensitive to peptide purity. Contaminants or aggregated protein can activate different TLR subtypes, creating confounding cytokine profiles that obscure the true thymosin alpha-1 signaling pathway.
Downstream Immune Cell Programming
The transcription factor activation described above translates into functional changes in immune cell behavior. The most critical effect is dendritic cell maturation. The process by which immature dendritic cells transition from an antigen-capturing phenotype to an antigen-presenting phenotype. Thymosin alpha-1 signaling upregulates surface expression of CD80 and CD86 (costimulatory molecules required for T-cell activation) and CD83 (a marker of dendritic cell maturity). Without these surface markers, dendritic cells cannot effectively prime naïve T-cells.
The cytokine microenvironment created by thymosin alpha-1 signaling shifts T-cell differentiation toward Th1 phenotypes. Elevated IL-12 production from dendritic cells activates STAT4 signaling in naïve CD4+ T-cells, driving T-bet expression. The master transcription factor for Th1 commitment. Th1 cells produce IFN-γ and IL-2, which support cytotoxic T-lymphocyte (CTL) activity and natural killer (NK) cell function. This is mechanistically distinct from Th2-skewed immunity, which favors antibody production over cell-mediated cytotoxicity.
Clinical trials in chronic hepatitis B patients demonstrated that twice-weekly thymosin alpha-1 injections for 24 weeks increased CD8+ T-cell IFN-γ production by 2.3-fold compared to baseline. The same trials showed no significant increase in IL-4 or IL-10 (Th2 cytokines), confirming the pathway's selectivity for Th1 responses. The practical implication: thymosin alpha-1 signaling is particularly effective in contexts requiring viral clearance or tumor surveillance. Both of which depend on robust CTL activity.
| Signaling Component | Thymosin Alpha-1 Effect | Time to Peak Response | Clinical Relevance | Professional Assessment |
|---|---|---|---|---|
| TLR-2/TLR-9 Binding | Direct receptor engagement with 2.8 μM affinity | 15–30 minutes post-dose | Initiates immune recognition without pathogen presence | This is the critical 'danger signal' that activates dormant dendritic cells without triggering inflammatory damage |
| MyD88 Myddosome Formation | Recruits IRAK-4/IRAK-1 complex to activated TLRs | 30–45 minutes post-dose | Propagates signal from membrane to cytoplasm | The bottleneck step. If MyD88 is deficient, thymosin alpha-1 effects are almost entirely abolished |
| NF-κB Nuclear Translocation | 3.2-fold increase in nuclear NF-κB at 60 minutes | 45–90 minutes post-dose | Drives IL-2, IL-12, TNF-α transcription | The primary driver of Th1-skewed cytokine production. This is what shifts T-cell programming |
| IRF-7 Activation | 4.7-fold increase in IFN-α mRNA at 6 hours | 4–8 hours post-dose | Essential for antiviral interferon response | Particularly relevant for viral infections. This pathway is why thymosin alpha-1 is used in hepatitis B/C treatment |
| Dendritic Cell Maturation | CD80/CD86 expression increased 3.2-fold at 12 hours | 12–24 hours post-dose | Required for effective T-cell priming | Without mature dendritic cells, the adaptive immune response cannot engage. This is the functional output of all upstream signaling |
Key Takeaways
- Thymosin alpha-1 activates the immune signaling pathway by binding TLR-2 and TLR-9 on dendritic cells, initiating MyD88-dependent signal transduction within 15–30 minutes.
- The pathway diverges into NF-κB and IRF-7 branches, upregulating IL-2, IL-12, TNF-α, IFN-α, and IFN-β transcription with peak effects at 4–12 hours post-administration.
- Dendritic cell maturation markers (CD80, CD86, CD83) increase 3.2-fold by 12–24 hours, enabling effective T-cell priming and Th1 differentiation.
- The thymosin alpha-1 signaling pathway selectively enhances Th1 immune responses. Favoring cytotoxic T-lymphocyte and natural killer cell activity over antibody-mediated immunity.
- Clinical hepatitis B trials showed 2.3-fold increases in CD8+ T-cell IFN-γ production after 24 weeks of twice-weekly thymosin alpha-1 dosing, demonstrating sustained pathway activation.
What If: Thymosin Alpha-1 Signaling Pathway Scenarios
What If TLR Receptors Are Genetically Deficient?
Use alternative immune adjuvants that engage different pattern recognition receptors.
Individuals with TLR-2 or TLR-9 genetic polymorphisms show blunted responses to thymosin alpha-1 in clinical cohorts. One Italian study found that TLR-9 -1237T/C polymorphism carriers had 54% lower IFN-α induction compared to wild-type. Alternative peptides like LL-37 (which signals through FPRL-1) or vitamin D receptor agonists can partially compensate by activating parallel immune pathways.
What If Cytokine Production Is Already Elevated?
Monitor for cytokine storm risk and adjust dosing frequency.
Thymosin alpha-1 signaling amplifies existing immune activity. In patients with autoimmune conditions or active viral infections where baseline IL-6 and TNF-α are elevated, adding thymosin alpha-1 can push cytokine levels into pathological ranges. Pre-treatment cytokine panels (IL-6, TNF-α, IFN-γ) can guide dosing. If baseline levels exceed 3× upper limit of normal, reduce thymosin alpha-1 dosing to once weekly or hold until inflammation resolves.
What If Dendritic Cells Are Functionally Impaired?
Combine thymosin alpha-1 with GM-CSF to restore dendritic cell differentiation.
Chemotherapy, radiation, and chronic steroid use deplete functional dendritic cell populations. In these contexts, thymosin alpha-1 has receptors to bind but insufficient target cells to activate. Granulocyte-macrophage colony-stimulating factor (GM-CSF) drives monocyte-to-dendritic-cell differentiation. Combining 5mcg/kg GM-CSF with thymosin alpha-1 restores pathway responsiveness in immunosuppressed patients.
The Evidence-Based Truth About Thymosin Alpha-1 Signaling
Here's the honest answer: thymosin alpha-1's mechanism is receptor-mediated signal transduction. Not vague 'immune support.' The peptide binds specific TLRs, activates defined intracellular pathways, and produces quantifiable changes in cytokine transcription and immune cell phenotypes. This isn't speculative immunology. The pathway has been mapped at the protein level using co-immunoprecipitation, knockdown studies, and transcriptome analysis.
What this means practically: thymosin alpha-1 works through a dose-dependent, time-limited mechanism that requires functional TLR and MyD88 expression. It doesn't 'boost immunity' in a general sense. It specifically enhances Th1-mediated immunity while leaving Th2 and regulatory T-cell pathways largely unaffected. For research applications requiring precise immune modulation, understanding this pathway isn't optional. Researchers working with immune cell culture, viral challenge models, or tumor immunology protocols need peptides synthesized to exact specifications. Aggregated or oxidized thymosin alpha-1 loses TLR binding affinity and produces inconsistent signaling outcomes.
Suppliers like Real Peptides provide high-purity, research-grade thymosin alpha-1 with verified amino acid sequencing and low endotoxin levels. Critical for isolating the thymosin alpha-1 signaling pathway from confounding LPS-mediated TLR activation.
The thymosin alpha-1 signaling pathway represents one of the most well-characterized examples of peptide-mediated immune modulation in molecular immunology. The cascade from TLR binding through MyD88 activation to transcription factor engagement happens predictably and reproducibly when proper peptide quality and dosing protocols are followed. For researchers investigating immune signaling mechanisms or clinicians considering thymosin alpha-1 in immunotherapy contexts, the pathway's specificity is both its strength and its limitation. It does exactly what the molecular biology predicts, nothing more.
Frequently Asked Questions
How does thymosin alpha-1 activate immune cells at the molecular level?▼
Thymosin alpha-1 binds to TLR-2 and TLR-9 receptors on dendritic cells and macrophages, triggering MyD88-dependent signaling cascades. This recruits IRAK kinases and activates TRAF6, which then phosphorylates downstream targets including the IKK complex and IRF-7. The result is NF-κB and IRF-7 translocation to the nucleus, upregulating transcription of IL-2, IL-12, IFN-α, and IFN-β within 4–6 hours. This mechanism is distinct from general immune ‘boosting’ — it specifically enhances pattern recognition receptor signaling.
What is the difference between thymosin alpha-1 and thymosin beta-4 signaling pathways?▼
Thymosin alpha-1 signals through TLR-2/TLR-9 and MyD88 to activate immune transcription factors (NF-κB, IRF-7), driving cytokine production and dendritic cell maturation. Thymosin beta-4 operates through a completely different mechanism — it binds G-actin monomers to regulate cytoskeletal dynamics and signals through integrin receptors to promote cell migration and wound healing. The two peptides share a name but have no overlapping signaling pathways or functional roles.
Can thymosin alpha-1 signaling trigger autoimmune responses?▼
Thymosin alpha-1 enhances existing immune surveillance but does not independently initiate autoimmunity in healthy individuals. However, in patients with pre-existing autoimmune conditions (rheumatoid arthritis, lupus, multiple sclerosis), amplifying Th1 cytokine production can exacerbate disease activity. Clinical monitoring of inflammatory markers (CRP, ESR, IL-6) is recommended when using thymosin alpha-1 in autoimmune-prone populations. The peptide activates danger signal pathways without providing the second signals required for true autoimmune T-cell activation.
How long does thymosin alpha-1 signaling persist after a single dose?▼
Peak signaling occurs 4–12 hours post-administration, with NF-κB translocation peaking at 60–90 minutes and cytokine transcription peaking at 6–8 hours. Functional effects on dendritic cell maturation (CD80/CD86 upregulation) persist for 48–72 hours. However, sustained immune modulation requires repeated dosing — clinical protocols typically use twice-weekly injections because single-dose effects return to baseline within 5–7 days.
What happens if MyD88 is genetically deficient?▼
MyD88 deficiency almost entirely abolishes thymosin alpha-1 signaling. Studies in MyD88 knockout mice showed no increase in IL-12 or IFN-α production following thymosin alpha-1 administration, and dendritic cell maturation markers remained unchanged. Human MyD88 deficiency is rare but documented — these individuals rely on TRIF-dependent TLR pathways (TLR-3, TLR-4) for immune function, which thymosin alpha-1 does not engage. Alternative immune adjuvants targeting different pathways are required.
Does thymosin alpha-1 affect Th2 or regulatory T-cell pathways?▼
Thymosin alpha-1 signaling selectively enhances Th1 responses with minimal direct effect on Th2 (IL-4, IL-5, IL-13) or Treg (IL-10, TGF-β) pathways. Clinical studies show no significant increase in IL-4 or IL-10 production following thymosin alpha-1 treatment. The selectivity results from the specific cytokine profile it induces — IL-12 and IFN-α directly promote Th1 differentiation while suppressing Th2 commitment. This makes thymosin alpha-1 poorly suited for antibody-mediated immunity or allergy contexts.
Can thymosin alpha-1 signaling be blocked or reversed?▼
Yes, TLR-2 and TLR-9 antagonists can block thymosin alpha-1 signaling at the receptor level. Additionally, NF-κB inhibitors (like BAY 11-7082) and JAK-STAT inhibitors (tofacitinib, baricitinib) downstream of cytokine receptors can blunt the functional effects. Clinically, discontinuing thymosin alpha-1 results in signal decay within 48–72 hours as activated transcription factors return to baseline and cytokine mRNA half-lives expire. There is no long-term ‘residual activation’ once dosing stops.
How does peptide purity affect thymosin alpha-1 signaling pathway activation?▼
Impurities — particularly aggregated peptide or residual synthesis reagents — can activate off-target TLRs or introduce endotoxin contamination that triggers non-specific inflammation. Research-grade thymosin alpha-1 should have >98% purity by HPLC and <1 EU/mg endotoxin to isolate the true thymosin alpha-1 signaling pathway. Aggregated or oxidized thymosin alpha-1 loses TLR-2 binding affinity, reducing dendritic cell activation by 40–60% compared to monomeric peptide.
What role does thymosin alpha-1 signaling play in viral infections?▼
Thymosin alpha-1’s IRF-7 activation pathway is particularly relevant for antiviral immunity. IRF-7 drives Type I interferon production (IFN-α, IFN-β), which establishes an antiviral state in surrounding cells and enhances NK cell and CTL activity. Clinical trials in chronic hepatitis B and C showed improved viral clearance rates when thymosin alpha-1 was combined with antiviral drugs — the mechanism is enhanced immune surveillance of infected hepatocytes, not direct antiviral activity.
Does thymosin alpha-1 signaling differ between immune cell types?▼
Yes — the pathway is most active in dendritic cells and macrophages, which express high levels of TLR-2 and TLR-9. T-cells and B-cells express lower TLR levels and show minimal direct response to thymosin alpha-1. The primary effect on T-cells is indirect: thymosin alpha-1 conditions dendritic cells to produce Th1-polarizing cytokines, which then program naïve T-cells during antigen presentation. Plasmacytoid dendritic cells show the strongest IRF-7 response due to constitutive IRF-7 expression.











