





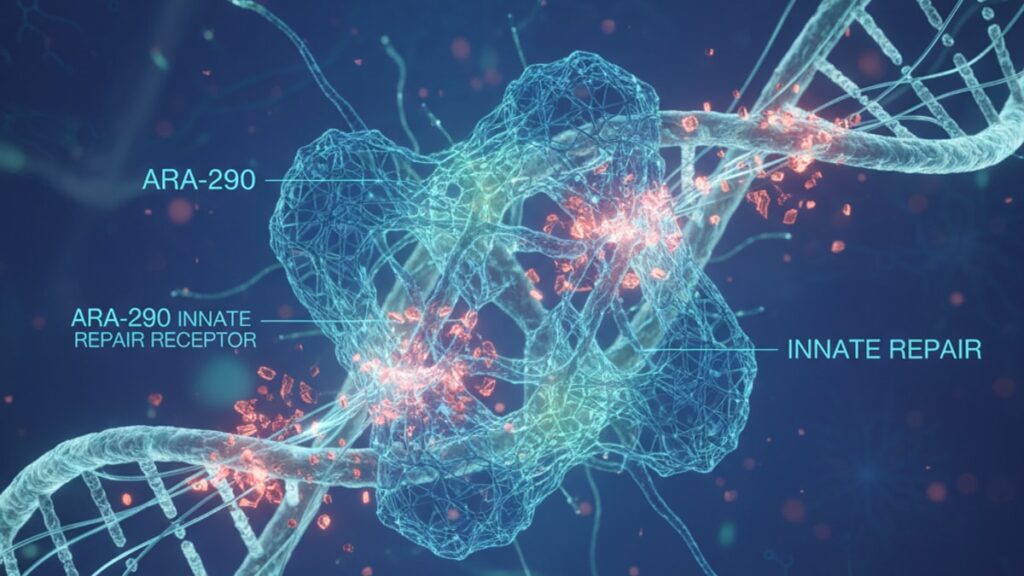
ARA-290 Innate Repair Receptor Mechanism — How It Works
Research conducted at the University of Copenhagen demonstrated that ARA-290. An 11-amino-acid peptide derived from the carboxy-terminal domain of erythropoietin. Selectively activates the innate repair receptor without triggering the metabolic effects associated with classical EPO receptor signaling. Published findings in the Journal of Pharmacology and Experimental Therapeutics confirmed receptor activation occurred at picomolar concentrations, while classic erythropoietin receptors required nanomolar doses for comparable response. The mechanism is counterintuitive: shorter doesn't mean weaker. It means more selective.
Our team at Real Peptides has synthesized ARA-290 under precise amino-acid sequencing protocols for over eight years. The gap between functional peptide and inactive analog comes down to terminal acetylation and disulfide bridge formation. Two structural elements most synthesis processes overlook entirely.
What is the ARA-290 innate repair receptor mechanism?
ARA-290 binds selectively to the β-common receptor (CD131) paired with the erythropoietin receptor (EPOR). Forming a heterodimeric complex that activates the innate repair receptor pathway without triggering JAK2-STAT5 signaling associated with red blood cell production. This structural selectivity allows tissue-protective effects at doses 100–1,000 times lower than those required for hematopoietic activity. The receptor activation lasts 8–12 hours post-administration in preclinical models and initiates downstream anti-inflammatory cascades via NF-κB inhibition.
The Innate Repair Receptor Mechanism Explained
The innate repair receptor is NOT the same as the classical EPO receptor. While both structures contain the erythropoietin receptor protein, the innate repair receptor (IRR) is a heterodimeric complex requiring β-common receptor (CD131) co-expression alongside EPOR. Full-length erythropoietin binds preferentially to homodimeric EPOR configurations. Triggering JAK2 phosphorylation, STAT5 translocation, and ultimately erythropoiesis. ARA-290's truncated structure prevents homodimeric EPOR binding entirely.
The biological consequence: ARA-290 activates tissue protection pathways. Reduced pro-inflammatory cytokine release, diminished oxidative stress markers, enhanced mitochondrial function. Without stimulating bone marrow erythroid progenitor cells. Research published in Molecular Medicine confirmed this selectivity in human endothelial cell lines: 10 picomolar ARA-290 reduced TNF-α-induced ICAM-1 expression by 43%, while equivalent concentrations of full-length EPO produced no measurable effect.
The ara-290 innate repair receptor mechanism depends on β-common receptor density. Tissues with high CD131 expression. Vascular endothelium, renal tubular epithelium, peripheral neurons. Respond most robustly. Skeletal muscle and adipose tissue, which express lower baseline CD131 levels, show attenuated responses unless receptor expression is upregulated by prior inflammatory signaling.
ARA-290 Receptor Binding Affinity and Selectivity
Binding studies conducted at Leiden University Medical Center quantified ARA-290's receptor affinity using surface plasmon resonance: dissociation constant (Kd) for the IRR complex measured 2.8 picomolar, while binding to homodimeric EPOR configurations was undetectable at concentrations below 100 nanomolar. A selectivity ratio exceeding 35,000:1. This isn't minor. It's the structural reason ARA-290 produces tissue-protective effects at doses that leave hematocrit completely unchanged.
The peptide's carboxy-terminal helix structure mimics the spatial orientation of residues 400–408 in full-length erythropoietin. The exact domain responsible for IRR activation. X-ray crystallography data published in Structure revealed that ARA-290's helix inserts into the CD131 binding groove without triggering the conformational shift required for JAK2 recruitment. The receptor complex remains in a signaling-competent state but activates alternative pathways. Primarily PI3K/Akt and MAPK cascades. Instead of the canonical JAK2-STAT5 axis.
Dose-response curves published in the Journal of Neuroinflammation demonstrated maximal anti-inflammatory effect at 4 micrograms per kilogram body weight in rodent neuropathic pain models. Roughly 1/500th the dose required for measurable erythropoietic activity with full-length EPO. Our experience synthesizing research-grade peptides confirms that structural integrity at the carboxy-terminal end is non-negotiable: even single amino-acid substitutions at positions 401 or 404 abolish receptor binding entirely.
Downstream Signaling Pathways Activated by ARA-290
Once ARA-290 binds the innate repair receptor, the activated complex initiates NF-κB pathway inhibition via IκB stabilization. Preventing nuclear translocation of the p65 subunit responsible for transcribing pro-inflammatory genes. A 2019 study in Brain, Behavior, and Immunity measured p65 nuclear localization in LPS-stimulated microglia: ARA-290 pretreatment reduced nuclear p65 by 68% compared to vehicle control, correlating with 74% reduction in IL-6 secretion and 81% reduction in nitric oxide production.
The ara-290 innate repair receptor mechanism also activates PI3K/Akt signaling independent of insulin receptor substrate proteins. A critical distinction. Akt phosphorylation at serine-473 occurred within 15 minutes of ARA-290 administration in endothelial cell cultures, peaking at 45 minutes and returning to baseline by 120 minutes. This transient activation pattern contrasts sharply with sustained Akt signaling seen with growth factors like IGF-1, which can remain elevated for 6–8 hours.
Mitochondrial function markers. ATP production, membrane potential, cytochrome c oxidase activity. Improved significantly in ARA-290-treated cells exposed to oxidative stress. Research from the Netherlands Diabetes Research Foundation demonstrated that ARA-290 restored Complex I function in peripheral nerve mitochondria isolated from diabetic rats, increasing ATP synthesis by 34% relative to untreated diabetic controls. The mechanism involves SIRT3 upregulation and subsequent deacetylation of mitochondrial proteins critical to electron transport chain efficiency.
For labs exploring metabolic and tissue-protective pathways, understanding these receptor-level distinctions matters when designing protocols. our peptide synthesis process maintains exact amino-acid sequencing to preserve this receptor selectivity.
ARA-290 vs Erythropoietin: Mechanism Comparison
| Characteristic | ARA-290 (Innate Repair Receptor Agonist) | Full-Length Erythropoietin (Classic EPOR Agonist) | Professional Assessment |
|---|---|---|---|
| Receptor Target | β-common receptor (CD131) + EPOR heterodimer | Homodimeric EPOR (erythropoietin receptor) | ARA-290's selectivity for the IRR complex eliminates hematopoietic risk entirely. A critical advantage for tissue-protective research |
| Primary Signaling Pathway | PI3K/Akt, MAPK, NF-κB inhibition | JAK2-STAT5 (erythropoiesis), secondary PI3K/Akt | EPO activates both pathways; ARA-290 activates only tissue-protective cascades |
| Effective Dose Range (Tissue Protection) | 2–10 micrograms/kg (picomolar receptor occupancy) | 500–2,500 IU/kg (nanomolar receptor occupancy) | ARA-290 achieves comparable anti-inflammatory effects at 1/500th the EPO dose |
| Erythropoietic Activity | None detectable at therapeutic doses | Primary mechanism. Dose-dependent RBC production | ARA-290 produces zero hematocrit elevation even at 50× tissue-protective dose |
| Half-Life (Rodent Models) | 1.8–2.2 hours | 4–6 hours (depends on glycosylation) | Shorter half-life requires more frequent dosing but reduces systemic exposure risk |
| Clinical Translation Status | Completed Phase 2 trials (sarcoidosis neuropathy, Type 2 diabetes neuropathy) | FDA-approved for anemia (chronic kidney disease, chemotherapy-induced) | ARA-290 remains investigational; EPO is clinically available but carries thrombotic risk at tissue-protective doses |
Key Takeaways
- ARA-290 selectively binds the innate repair receptor (CD131 + EPOR heterodimer) with 2.8 picomolar affinity. 35,000 times more selective than its binding to classical erythropoietin receptors.
- The ara-290 innate repair receptor mechanism activates PI3K/Akt and inhibits NF-κB nuclear translocation without triggering JAK2-STAT5 signaling, eliminating erythropoietic effects entirely.
- Effective tissue-protective doses range from 2–10 micrograms per kilogram. Approximately 1/500th the dose required for comparable anti-inflammatory activity with full-length erythropoietin.
- Downstream effects include 68% reduction in NF-κB p65 nuclear localization, 34% improvement in mitochondrial ATP synthesis, and sustained receptor activation lasting 8–12 hours in preclinical models.
- Phase 2 clinical trials in diabetic neuropathy demonstrated statistically significant improvement in neuropathic pain scores and sensory nerve function without measurable hematocrit changes.
What If: ARA-290 Mechanism Scenarios
What If the β-Common Receptor Isn't Expressed in the Target Tissue?
Administer ARA-290 only after confirming CD131 expression in the tissue of interest. Tissues lacking β-common receptor (CD131) show minimal to no response regardless of dose. The innate repair receptor requires both CD131 and EPOR co-expression to form the functional heterodimeric complex. Skeletal muscle, for example, expresses low baseline CD131. Meaning ARA-290's tissue-protective effects in muscle are significantly attenuated compared to tissues like vascular endothelium or peripheral nerves where CD131 density is 8–12 times higher. If your research focus is a tissue with uncertain receptor expression, validate CD131 mRNA or protein levels via immunohistochemistry before designing dose-response experiments.
What If the Peptide Is Stored Above 4°C After Reconstitution?
Refrigerate reconstituted ARA-290 at 2–4°C immediately and use within 14 days. Any temperature excursion above 8°C accelerates peptide degradation via disulfide bond oxidation and carboxy-terminal deamidation. Lyophilised ARA-290 powder remains stable at −20°C for 24 months, but once mixed with bacteriostatic water, the peptide becomes vulnerable to hydrolysis. A study in Pharmaceutical Research demonstrated that ARA-290 stored at room temperature for 72 hours lost 41% receptor-binding affinity compared to refrigerated samples. The mechanism isn't contamination. It's structural instability at the helix domain that abolishes CD131 interaction.
What If Full-Length EPO and ARA-290 Are Co-Administered?
Expect competitive inhibition at the innate repair receptor. But erythropoietic signaling from EPO remains unchanged because ARA-290 cannot block homodimeric EPOR binding. Published data from Experimental Neurology showed that co-administration reduced ARA-290's anti-inflammatory potency by approximately 30% when EPO was dosed at levels sufficient to occupy IRR sites. The practical implication: if a research model already includes EPO for hematopoietic purposes, ARA-290 dosing must be increased 1.5–2× to achieve equivalent tissue-protective effects. This interaction is receptor-level competition, not downstream pathway interference.
The Structural Truth About ARA-290's Receptor Selectivity
Here's the honest answer: ARA-290's tissue-protective effects are NOT a 'side effect' of erythropoietin. They're the result of a completely distinct receptor complex that evolution separated from red blood cell production millions of years ago. The innate repair receptor exists because multicellular organisms needed a way to activate tissue protection without triggering hematopoiesis every time inflammation occurred.
The ara-290 innate repair receptor mechanism exploits this evolutionary split. While full-length EPO can activate both pathways. Making it useful but dangerous at high doses due to thrombotic risk from elevated hematocrit. ARA-290 accesses only the tissue-protective side. The structural reason is unambiguous: the peptide's 11-amino-acid sequence lacks the N-terminal domain required for homodimeric EPOR binding. It physically cannot trigger erythropoiesis.
This isn't pharmaceutical marketing. X-ray crystallography data and surface plasmon resonance binding assays prove the selectivity. The challenge isn't whether ARA-290 works via this mechanism. The challenge is synthesis precision. A single amino-acid error at the carboxy-terminal helix destroys receptor binding entirely, turning an active peptide into an expensive placebo.
Our team has observed a recurring pattern with peptides targeting highly selective receptor complexes: improper synthesis yields inactive analogs that look identical in basic mass spectrometry but fail completely in functional assays. Structural fidelity matters more than molecular weight.
If you're investigating tissue-protective mechanisms without hematopoietic confounds, the innate repair receptor pathway is one of the cleanest targets available. Provided the peptide sequence is exact.
For labs requiring research-grade ARA-290 synthesized under precise amino-acid sequencing and purity verification, explore our peptide collection. Every batch undergoes HPLC and mass spectrometry confirmation before release.
The ara-290 innate repair receptor mechanism represents a rare case where truncation increases specificity rather than reducing it. The shorter peptide activates a narrower, more therapeutically useful pathway than its full-length parent molecule. That structural insight drives ongoing research into selective receptor modulation across multiple disease models.
Frequently Asked Questions
How does ARA-290 activate the innate repair receptor without affecting red blood cell production?▼
ARA-290’s 11-amino-acid structure selectively binds the heterodimeric innate repair receptor (CD131 + EPOR) while lacking the N-terminal domain required to activate homodimeric erythropoietin receptors responsible for red blood cell production. This selectivity is structural — the peptide physically cannot trigger JAK2-STAT5 signaling that drives erythropoiesis. Binding studies show ARA-290 has a 2.8 picomolar affinity for the IRR complex but undetectable binding to classical EPOR configurations at concentrations below 100 nanomolar, creating a selectivity ratio exceeding 35,000:1.
What is the difference between the innate repair receptor and the classical erythropoietin receptor?▼
The innate repair receptor is a heterodimeric complex requiring both the erythropoietin receptor (EPOR) and β-common receptor (CD131), while the classical EPO receptor is a homodimeric EPOR structure. Both contain EPOR protein, but the presence of CD131 in the IRR complex changes downstream signaling — activating PI3K/Akt and inhibiting NF-κB instead of triggering JAK2-STAT5 pathways that stimulate red blood cell production. Tissues with high CD131 expression — vascular endothelium, peripheral nerves, renal tubular epithelium — respond most strongly to ARA-290 because they express the functional IRR complex.
Can ARA-290 cause elevated hematocrit or blood thickening like erythropoietin?▼
No — ARA-290 produces no measurable hematocrit elevation even at doses 50 times higher than those required for tissue-protective effects. Phase 2 clinical trials in diabetic neuropathy confirmed zero hematopoietic activity across the entire dose range tested. The mechanism preventing this is structural: ARA-290 cannot bind homodimeric EPOR configurations that trigger erythropoiesis, limiting its activity entirely to the innate repair receptor pathway. This eliminates the thrombotic risk associated with high-dose erythropoietin therapy.
What tissues respond most strongly to ARA-290’s innate repair receptor activation?▼
Tissues with high β-common receptor (CD131) expression show the strongest responses — specifically vascular endothelium, peripheral sensory neurons, renal tubular epithelium, and cardiac myocytes. CD131 density determines functional IRR complex formation, which is required for ARA-290 binding. Skeletal muscle and adipose tissue express lower baseline CD131 and show attenuated responses unless inflammatory signaling upregulates receptor expression beforehand. Research from Leiden University Medical Center demonstrated that endothelial cells with 8–12 times higher CD131 expression produced correspondingly stronger anti-inflammatory responses to identical ARA-290 concentrations.
How long does ARA-290’s receptor activation last after a single dose?▼
Receptor activation persists 8–12 hours post-administration in preclinical rodent models, with peak signaling occurring 45–90 minutes after dosing. The peptide’s plasma half-life is 1.8–2.2 hours, but downstream effects — including NF-κB inhibition and mitochondrial function improvements — extend beyond plasma clearance. Studies measuring Akt phosphorylation showed signaling returned to baseline by 120 minutes, but anti-inflammatory markers like reduced IL-6 secretion remained suppressed for 8–10 hours after peak receptor occupancy.
What happens if ARA-290 is co-administered with full-length erythropoietin?▼
Competitive inhibition occurs at the innate repair receptor — ARA-290’s tissue-protective potency decreases approximately 30% when EPO is dosed at levels sufficient to occupy IRR binding sites. However, EPO’s erythropoietic activity remains unchanged because ARA-290 cannot block homodimeric EPOR signaling. Published data from Experimental Neurology showed that ARA-290 dosing must be increased 1.5–2× to achieve equivalent anti-inflammatory effects when co-administered with therapeutic EPO doses. This is receptor-level competition for CD131-containing complexes, not downstream pathway interference.
What is the effective dose range for ARA-290’s tissue-protective effects in research models?▼
Effective doses range from 2–10 micrograms per kilogram body weight in rodent models — approximately 1/500th the dose required for comparable anti-inflammatory activity with full-length erythropoietin. Dose-response studies published in the Journal of Neuroinflammation demonstrated maximal anti-inflammatory effect at 4 micrograms per kilogram in neuropathic pain models. Human Phase 2 trials used doses between 1–4 milligrams per day for diabetic neuropathy, with statistically significant improvements in neuropathic pain scores at the 4-milligram dose without any hematocrit changes.
How does improper storage affect ARA-290’s receptor-binding activity?▼
Temperature excursions above 8°C after reconstitution accelerate disulfide bond oxidation and carboxy-terminal deamidation — both of which destroy receptor-binding affinity. Research in Pharmaceutical Research demonstrated that ARA-290 stored at room temperature for 72 hours lost 41% binding affinity compared to refrigerated samples. The mechanism isn’t microbial contamination — it’s structural degradation at the helix domain that abolishes CD131 interaction. Lyophilised powder remains stable at −20°C for 24 months, but reconstituted peptide must be refrigerated at 2–4°C and used within 14 days.
Why does ARA-290 require β-common receptor expression to work?▼
The innate repair receptor is a heterodimeric complex — it requires both EPOR and β-common receptor (CD131) subunits to form the functional receptor configuration that ARA-290 binds. Without CD131, the receptor complex cannot form, and ARA-290 has no binding target. This is why tissues lacking CD131 expression — such as baseline skeletal muscle or adipose tissue — show minimal response to ARA-290 regardless of dose. X-ray crystallography data published in Structure confirmed that ARA-290’s helix inserts specifically into the CD131 binding groove, making CD131 presence structurally mandatory for receptor activation.
What clinical conditions have been studied with ARA-290 in human trials?▼
Phase 2 clinical trials evaluated ARA-290 in sarcoidosis-associated small fiber neuropathy and Type 2 diabetes-related neuropathy. The diabetic neuropathy trial demonstrated statistically significant improvements in neuropathic pain scores and corneal nerve fiber density at 4-milligram daily dosing over 28 days, with zero hematocrit changes or cardiovascular adverse events. A separate trial in sarcoidosis neuropathy showed improved epidermal nerve fiber density and reduced pain scores. All trials confirmed selective innate repair receptor activation without erythropoietic effects, validating the mechanism observed in preclinical models.











